
Littlewood JM. Honking [Letter] Lancet. 1(8182):1367, 1980 Jun 21. https://pubmed.ncbi.nlm.nih.gov/6104167/
Honking is a psychogenic cough tic in children; it is a troublesome complaint. The cough is a noisy bark or honking, repeated frequently while the child is awake, but absent during sleep. Clinical and laboratory findings are negative, and cough suppressants and other medications are ineffective. The cough usually starts in the winter months and may be preceded by an upper respiratory tract infection. School phobia is frequently a contributory cause, but other psychological problems must also be considered. Diagnostic feature is that it is not present during sleep.
Littlewood JM. The diagnosis of cystic fibrosis. [ Journal Article] Practitioner. 224(1341):305-7, 1980 Mar. https://pubmed.ncbi.nlm.nih.gov/7403012/
This paper represented a milestone with my lifetime involvement in the many aspects of cystic fibrosis. By 1974 I had been invited to be Chairman of the West Riding and District Branch of the Cystic Fibrosis Research Trust although I had only minimal clinical involvement at that time but by 1984 I had become their Medical Adviser and also a member of the UK CF Trust’s Research and Medical Advisory Committee.

This was the first article I had written on cystic fibrosis. I was invited to write it by Dr Archie Norman, a senior consultant paediatrician at Great Ormond Street and one of the leading paediatricians involved with the CF Research Trust from its foundation in 1964.

I had started neonatal screening for CF in 1975 at St Mary’s Maternity Hospital, in Leeds where I was responsible for the paediatric care of 3000 infants per year. As we started to detect the occasional infant with CF I started a small monthly clinic for them at Seacroft Hospital, my main base. Also in the late Seventies, we carried out our first research on our few CF children with members of Professor Monty Losowsky’s University Department of Medicine at St James’s (described below Congdon et al 1981). I think the dynamic Mr Ron Tucker, the Director of the CF Research Trust was a definite influence in getting me involved for the next three decades!
The early history of the Leeds Cystic Fibrosis Service is described in detail on my personal website (www.jimlittlewood.com) in two chapters – “Origins and Development of the Leeds Regional Cystic Fibrosis Service”. Parts 1 & 2. These are also on the Leeds CF website. (cystic fibrosis online.com)
Littlewood J M, Crollick Avril J, Richards IDG. Childhood coeliac disease is disappearing. Lancet 1980;ii:1359. https://pubmed.ncbi.nlm.nih.gov/6109168/
During the Seventies paediatric consultants in the Yorkshire Region increasingly referred children to Seacroft for Mr Alan Steel’s reliable sweat tests and our jejunal biopsy service which I had started in the late Sixties.
In 1968 I had obtained a paediatric sized Crosby intestinal biopsy capsule (Watson capsule) and started a paediatric jejunal biopsy service; this facility soon attracted referrals from paediatricians in the region to confirm or exclude coeliac disease in their children. We eventually changed to the twin port Kilby modification of the Crosby capsule to use one specimen for histology and the other for disaccharide estimation. In this venture of our biopsy service I had the invaluable help of my friend and colleague Dr. Sidney Smith, our paediatric radiologist, who was invaluable for rapid X-ray screening of the capsule and manoeuvring it from the stomach into the duodenum. Also , Dr. Mike Mason a consultant pathologist at St James’s for histology reports.

Later during the Seventies Dr. Avril Crollick was appointed as my Clinical Assistant and eventually performed many of the biopsies until I started using the paediatric endoscope for the purpose during the Eighties.
The ability to obtain jejunal mucosa for histological and biochemical examination from children was one of the major advances at time. Prof. John Walker-Smith considers this technique was the start of paediatric gastroenterology.
The characteristic histological small bowel appearance of subtotal villous atrophy of gluten induced coeliac disease was first identified in 1954 by Paulley, a physician in Ipswich, in laparotomy specimens from four
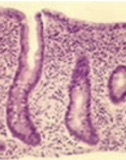

adults with idiopathic steatorrhoea (coeliac disease). In 1957 Margot Shiner was the first to obtain specimens by per oral small bowel biopsy from an 8-year old child; soon a series in children was reported by Charlotte Anderson.
Jejunal biopsy with a paediatric sized Crosby capsule was a relatively new technique in the Sixties. Initially Sir Wilfred Sheldon (the senior consultant paediatrician at Great Ormond Street (1901-1983)) and a senior registrar at GOS at the time, Dr. Eddie Tempany (1930-2010) who was subsequently Professor of Paediatrics in Dublin), initially considered the investigation to be “outside the scope of routine investigations in children” due to the increased difficulty and risk of perforation reported in small children. It is true there were a number of reports of intestinal perforation form various centres Certainly on one occasion, when I was working at GOS and observing Eddie performing a biopsy, the capsule could not be retrieved until it loosened its attachment to the mucosa the next day; but the child was fine.


However, my delay in starting jejunal biopsies in Leeds was mainly related to the fact that Sir Wilfred Sheldon and our Professor Stuart Craig were old friends and, as I was then only a lecturer in Prof Craig’s department, he did not approve of my performing jejunal biopsies. So I had to wait until 1968, when I became an NHS consultant, to start out paediatric jejunal biopsy service!

Over the next 25 years in my unit we had no serious complications. Subsequently elsewhere I have described in detail our experience with over 1000 jejunal biopsies and 108 children with coeliac disease in Professor Peter Howdle’s book (Howdle PD (ed) Coeliac Disease. Clinical Gastroenterology. Vol9/No2. 295-328. Bailliere Tindall, 1995).
Peter, a major friend of paediatrics, eventually succeeded Monty Losowsky as Professor of Medicine at St James’s. He was an excellent and helpful colleague in many areas of gastroenterology where his expertise benefited many of our young patients including those with cystic fibrosis, coeliac disease and inflammatory bowel disease.
To return to “Childhood Coeliac Disease is Disappearing”. During the previous eleven years from 1968 we (myself and Avril Crollick) had been providing a paediatric jejunal biopsy service for paediatric colleagues in Leeds and parts of our region of Yorkshire region who wished to use it for their infant and child patients. During this period 113 children were diagnosed as having coeliac disease. Approximately half these children were followed at my Seacroft clinic and from 1980 at the St James’s clinic, the remainder being under the care of the paediatricians who first referred them.

All the children had typical subtotal villous atrophy or severe partial villous atrophy and all had a definite clinical response to withdrawal of dietary gluten. Most with normal biopsies at follow-up were challenged with gluten powder to confirm a return of the villous atrophy. Those who failed to relapse (about 5% of patients) have NOT been included in the 113 patients reported here. Children less than 2 years old at diagnosis were challenged in their 3rd or 4th years to confirm the diagnosis.
The obvious fall in the number of children with coeliac disease was related to colleagues’ referral habits. Also hospital admission for coeliac disease in the Yorkshire region fell dramatically from 588 in 1978 to 102 in 1979; the majority of these admissions for coeliac disease were children.This is the first report of the falling incidence of coeliac disease in UK children and was possible thanks to our colleagues in the Yorkshire region referring their children for jejunal biopsy.

A number of changes in infant feeding practice in 1973 had followed DHHS advice on the later introduction of cereals, avoidance of hyperosmolar feeds and an increase in the incidence of breast feeding. We believed the incidence of coeliac disease was directly related to these changes in infant feeding practices in the mid-1970s. The increased prevalence of breast feeding, the consequent reduction in gastroenteritis and/or early immunological challenge from cow’s milk protein and the later exposure to gluten in the diet may all have been important factors in the observed change in incidence of coeliac disease.
We were grateful to Professor Gerald Richards of the University Department of Community Medicine and General Practice who help us show that the findings were significant.
Dandona P. Littlewood JM. Ramdial L. Evans R. Two tier screen for cystic fibrosis. [ Letter] Lancet. 1(8216):380, 1981 Feb 14 https://pubmed.ncbi.nlm.nih.gov/6110005/
I don’t remember t he precise details of this suggestion of a two tier screen for CF although I do remember discussing it with a very enthusiastic doctor at the Toronto CF meeting in 1980. I had a poster with our results on neonatal screening for CF with the BM Meconium test at that meeting and we had a discussion in relation to this – perhaps some way of reducing the false positives which were a concern to some. Our results were published and are discussed below (Evans RT et al, 1981 PMID: 7276211)

On searching for information I discover that Dr Paresh Dandona by 2020 has 613 articles listed on PubMed !! This letter is reference number 577. He is the SUNY Distinguished Professor, Department of Medicine Jacobs School of Medicine and Biomedical Sciences. He specialises in Endocrinology, Diabetes and Metabolism and in 2020 received the Madras Diabetes Research Foundation Lifetime Contribution Award. “One of the world’s leading experts in the treatment of diabetes and vascular disease, Dandona’s work on diabetes and metabolic disorders includes leading clinical trials for new treatments to exploring basic scientific mechanisms involved in both Type 1 and Type 2 diabetes”.
Congdon P. Mason MK. Smith S. Crollick A. Steel A. Littlewood J. Small-bowel mucosa in asymptomatic children with celiac disease. Mucosal changes with gluten-free diets. [Journal Article] American Journal of Diseases of Children. 135(2):118-21, 1981 Feb https://pubmed.ncbi.nlm.nih.gov/7468543/
Even though children with coeliac disease appear to be progressing satisfactorily while taking gluten free diets, dietary lapses and persisting mucosal abnormalities are common. Of 32 children with coeliac disease proven by biopsy only ten kept regularly to their diet, and 11 continued to ingest gluten regularly.
Despite satisfactory clinical progress, intestinal biopsy specimens after at least one year of a gluten free diet were markedly abnormal in 8 and normal in only 14 children.
Laboratory criteria available at the time failed to detect those with persisting mucosal abnormalities. These included serum and RBC folate, immunoglobulins, I-hour blood xylose. So clinical and laboratory criteria did not accurately detect those children with persistent mucosal abnormalities. The dietitian will usually be able to detect gluten ingestion but this assessment does not accurately differentiate between those with mild and severe villous atrophy.


If persistent mucosal damage is harmful over a long period, the only way at that time to ensure that the small-bowel mucosa returns to normal and remains so was by intestinal biopsy. Of 32 biopsy specimens, only 14 were normal or mildly abnormal (figure 1). Ten showed mild to moderate degree of partial villous atrophy (figure 2); three showed severe partial villous atrophy (figure 3); five showed subtotal villous atrophy (figure 4).
There are now (in 2026) a number of blood tests such as the Transglutaminase Antibodies (tTG-IgA) and a number of others (total serum IgA and deaminated gliadin peptide).
Congdon PJ. Kelleher J. Edwards P. Littlewood JM. Benign carotenaemia in children. [ Case Reports. Journal Article] Archives of Disease in Childhood. 56(4):292-4, 1981 Apr. https://pubmed.ncbi.nlm.nih.gov/7247441/

During the 1970s I was asked to see a young child who was noted to be slightly, but quite definitely, yellow first noted soon after a transatlantic air flight. I initially thought of the effects of altitude on children with thalassaemia and the haemolysis they may experience. Yet this child appeared very well although he was quite obviously yellow in the face, particularly in the nasolabial folds, but interestingly, there was no yellow discoloration of the whites of his eyes.Full physical examination was normal as were the initial full blood count and liver function tests. However, at the time we were fortunate that Dr Jerry Kelleher, in the Department of Medicine at St James University Hospital, Leeds was a world expert on vitamins. He was prepared to estimate the blood carotene level and this was markedly elevated at 510 ug/100ml. In our laboratory levels in healthy children were 105+-27ug/100ml (mean+-SD)
Once I was aware of this condition of hypercarotenaemia I soon recognised five other infants and young children with high levels of carotene but who were otherwise very well. So we termed the condition “Benign Carotenaemia” and published this report of six children where the initial serum carotene levels of 3 infants ranged from 320 to 510 ug/100ml (Normal <40-99 ug/100 ml)
We suggested that the cause was a high intake of dietary carotene made available by the frequent use of carrots in proprietary infant weaning foods. Communication with the H J Heinz Company in 1980 confirmed that carrots are cheap and readily available and were added to most varieties of their strained savoury baby foods. Also the crushing and processing of the carrots in the foods was likely to enhance the release of the carotene.
To this day I still notice infants who are obviously yellow and undoubtedly must have benign carotenaemia, but it does not seem to do them any harm and usually goes undiagnosed. I have included the illustration of a similar child from the internet (figure from DermaAmin) who shows obvious yellow skin discolouration, particularly of the nose, when compared with the father’s skin, but obviously has sparing of the sclera of the eyes.

The late Dr Peter Congdon (1944-1987) was a modest self-effacing and thoroughly excellent senior registrar with me at the time. He was involved in further publications from my unit in the early Eighties and was a great help when the 3000 neonates from St Marys and 2000 neonates in the old St James’s unit transferred to the new large neonatal unit in Gledhow Wing at St James’s in 1980. He had spent a year at the Hospital for Sick Children,Toronto in 1975 where he first encountered the higher technology of modern neonatal care. He was eventually appointed consultant neonatologist at the Leeds General Infirmary where he developed an excellent regional neonatal intensive care service. His premature death from cancer in 1987 was a great tragedy
Blumenthal I. Kelleher J. Littlewood JM. Recurrent abdominal pain and lactose intolerance in childhood. [Journal Article] British Medical Journal Clinical Research Ed.. 282(6281):2013-4, 1981 Jun 20. https://pubmed.ncbi.nlm.nih.gov/6788173/
Dr Ivan Blumenthal was my senior registrar at this time. He was originally from South Africa and a very pleasant and competent doctor. At the time opinions varied as to the frequency with which lactose intolerance was a cause of significant recurrent abdominal pain in children – estimates varied from over 25% to only 2%.
Ian Blumenthal studied 26 Caucasian children who had significant recurrent abdominal pain using lactose hydrogen breath tests. This is a test relying on undigested lactose passing through to the colon where it is fermented and produces excessive hydrogen in the patient’s breath.
Results indicating lactose malabsorption were obtained in only the three of the 26 children, two of whom experienced significant pain after ingesting lactose, one of whom improved on a lactose free diet.
We concluded lactose intolerance to be an infrequent cause of abdominal pain in Caucasian children and should not be diagnosed on a positive lactose breath test alone but only when accompanied symptoms induced by the lactose ingestion and subsequent relief from removal of lactose from the diet.
This was a nice clear and practically useful result as recurrent abdominal pain is a common problem in children referred to the paediatric outpatients and often considered to be an emotional problem. Yet another study where we combined with Jerry Kelleher of the University Department of Medicine at St James’s for the hydrogen breath tests.
Evans RT. Little AJ. Steel AE. Littlewood JM. Satisfactory screening for cystic fibrosis with the BM meconium procedure. [ Journal Article] Journal of Clinical Pathology. 34(8):911-3, 1981 Aug. https://pubmed.ncbi.nlm.nih.gov/7276211/

St Mary’s Maternity Hospital in Leeds was initially the Bramley Union Workhouse which opened in 1871. By the time I was appointed a NHS consultant in 1968 one part of my job was to provide paediatric cover there. By this time it had become the largest maternity unit in the city but with a slightly less privileged clientele than the other two maternity units in the city – the Leeds Maternity Hospital, Hyde Terrace and St James’s University Hospital. A significant proportion of our 3000 annual maternal admissions were for social reasons. It was slightly “out on a limb” and not really ideal for acute obstetrics but the staff were very dedicated and there were plenty of babies and a really excellent special care baby unit sister, Doreen Burton.
My personal interest in newborn screening had developed some years previously during the mid-Sixties when I was lecturer in paediatrics with Professor Craig at the Leeds Maternity Hospital. This eventually led to an MD thesis in 1968 on the incidence of neonatal urinary tract infection and various other related publications from the thesis (most are reviewed in this Paper trail).
Although from 1968 my role as a general paediatrician included the paediatric supervision of the 3000 infants born each year at St Mary’s, neonatology in the days before prolonged ventilation of premature neonates developed during the Seventies was quite different and much less demanding than today. In the mid-Seventies, after discussion with Miss Little, the very supportive Matron, it was agreed we introduce the recently available BM Meconium screening test for cystic fibrosis. The BM meconium test depends on the abnormally high protein content of the meconium (first bowel action) of infants with cystic fibrosis.

Wiser’s study (figure) using immunoelectrophoresis, the meconium from five infants with a history of CF was examined for increased protein. Three infants with CF (1,2,4) had obviously raised albumin levels, which did not occur in the two unaffected but “at risk of CF” infants (3,5) or in two healthy controls (6,7)
Later Schutt & Isles (1968) from Bristol showed that the increased meconium albumin could be recognised very simply by using the Albustix dipstick test designed for testing urine for albumin. A few drops of water mixed

with a small amount of meconium was placed on a tile and an Albustix applied to the edge of the drop when it would turn blue if there was excess albumin (figure of urine Albustix). This method eventually formed the basis of the commercially available Boehringer Mannheim test (BM test) used with variable success in a number of neonatal CF screening studies in the Seventies (Stephan et al, 1975; Ryley et al, 1979; Evans et al, 1983).
– Schutt WH, Isles TE. Protein in meconium from meconium ileus. Arch Dis Child. 1968 Apr;43(228):178–181.Â
– Stephan U, Busch EW, Kollberg H, Hellsing K. Cystic fibrosis detection by means of a test-strip. Pediatrics. 1975 Jan; 55(1):35–38
– Ryley HC, Neale LM, Brogan TD, Bray PT. Screening for cystic fibrosis by analysis of meconium for albumin and protease inhibitors. Clin Chim Acta. 1975 Oct 15;64(2):117–125.
So in 1975 we introduced neonatal screening for CF at St Mary’s Maternity Hospital in Leeds. After a few weeks on my frequent visits to the wards I frequently noted little plastic pots containing small quantities of meconium on the window sills, some even growing mould!! It soon became quite obvious that even this simple strip test was asking too much of the overworked but always willing and interested midwives! Fortunately Dr. Robert Evans, at the time the consultant chemical pathologist across the city at St James University Hospital, agreed to perform our BM tests in his laboratory on specimens of meconium collected into tiny pots by the midwives from all infants. This system worked very well and in 1980 we already had a poster which I presented at the Toronto CF Conference and in 1981 we published our favourable results since we started BM screening in 1975 (Evans et al, 1981). Although by 1981 most paediatricians had either never started or by this time already rejected the BM test for neonatal CF screening, at St Mary’s Maternity Hospital in East Leeds the test had become an established routine.
We had more success than in many other places and our false negative rate of only 12% – but, as described above, I found, as had the people in Veneto Italy, that the test must to be carried out in the laboratory with proper quality control and not on the wards by the midwives who, we soon realised, were far too busy. This was undoubtedly the reason other studies had failed.
Neonatal CF screening became an established routine at St Mary’s, Leeds Although others were reporting the BM test was unsatisfactory (Stephan et al ,1975; Ryley et al 1979; Prosser et al 1974), it slowly became apparent that we were obtaining reasonable results, but only because the test was performed in the laboratory with proper quality control which was not the case elsewhere.

Dr Henry Riley was a bacteriologist working in Cardiff. He was involved with a number of the early screening studies and had a long term interest in CF with the European Cystic Fibrosis Society. Even up to 2023 he provided a regular list of the recent references to cystic fibrosis.
The other important reason many paediatricians were unenthusiastic about neonatal CF screening was they did not believe early diagnosis affected the long-term outlook; of course this was true if early diagnosis was not accompanied by early effective treatment as ws the case in the UK. Even Professor Peter Phelan, the distinguished Australian paediatric pulmonologist, in 1982 questioned the value of early diagnosis. It is true that in the Seventies and Eighties there was no published evidence that early diagnosis through neonatal screening and the start of treatment was available at the time, improved the eventual appalling outlook for infants with CF. However, in my opinion common sense and clinical observations (at times a better guide than systematic reviews!), suggested otherwise! However, it was not until 2001 that definite generally accepted evidence of long term benefit on growth was published from Phillip Farrell’s unit in Wisconsin (Farrell et al, 2001). In fact, a large study from Wales and West Midlands in the Eighties, funded by the Cystic Fibrosis Trust had failed to show an advantage for the screened infants (Chatfield et al 1991). This was almost certainly due to the fact that many of the CF infants identified did not receive specialist CF centre care but were treated at a number of local hospitals.
– Prosser R, Owen H, Bull F, Parry B, Smerkinich J, Goodwin HA, Dathan J. Screening for cystic fibrosis by examination of meconium. Arch Dis Child. 1974 Aug;49(8):597–601.Â
– Chatfield S, Owen G, Ryley HC, Williams J, Alfaham M, Goodchild MC, Weller P. Neonatal screening for cystic fibrosis in Wales and the West Midlands: clinical assessment after five years of screening. Arch Dis Child. 1991 Jan;66(1 Spec No):29-33.Â
– Farrell PM, Kosorok MR, Rock MJ, Laxova A, Zeng L, Lai HC, Hoffman G, Laessig RH, Splaingard ML. Early diagnosis of cystic fibrosis through neonatal screening prevents severe malnutrition and improves long-term growth. Wisconsin Cystic Fibrosis Neonatal Screening Study Group. Pediatrics. 2001 Jan; 107(1):1-13.
3So despite the lack of national and local enthusiasm (including both cwwonsultant paediatricians at the other two maternity units in Leeds) neonatal CF screening became an established routine in our own neonatal unit at St Mary’s, Bramley, Leeds. In the first 5 years from 1975, 15,734 newborns were screened; a positive BM Test was obtained in 130 of whom 7 had CF. This was the expected incidence of 1/2243 live births. Also the false positive rate was an acceptable 0.83% (Evans et al, 1981).
– Evans RT, Little AJ, Steel AE, Littlewood JM. Satisfactory screening for cystic fibrosis with the BM meconium procedure. J Clin Pathol 1981;34(8):911-3. https://pubmed.ncbi.nlm.nih.gov/7276211/
Periodically during this first five years from 1975 we would identify a newborn with CF and it would seem sensible to start treatment with a regular anti-staphylococcal antibiotic (cloxacillin) and also start physiotherapy, pancreatic enzymes and vitamin supplements as recommended by Dr. David Lawson one of the UK’s few leading experts at the time.
We used the BM test for the next 20 years in the Eastern half of Leeds both at St Mary’s and later also at St James’s University Hospital when the two units merged and moved into new accommodation at St James’s in 1980. Dr. Ian Forsythe, the consultant paediatrician in charge of the St James’s neonatal unit before we merged in 1980, reluctantly agreed to introduce the test; he usually voiced his public opposition to CF screening at every opportunity! We eventually replaced the BM test with the immunoreactive trypsin heel prick test (IRT) in 1995 at which time the

neonatal unit at the Leeds General Infirmary eventually agreed to introduce neonatal CF screening. So there has been continuous neonatal CF screening in East Leeds first at St Mary’s and later at St James’s since 1975. Only one area of Italy had been screening continuously for longer having started in 1973/4 by Prof. Gianni Mastella at the Cystic Fibrosis Centre in Verona, Italy (Mastella et al, 2001).
– Mastella G1, Zanolla L, Castellani C, Altieri S, Furnari M, Giglio L, et al. Neonatal screening for cystic fibrosis: long-term clinical balance. Pancreatology. 2001;1(5):531-7.
In 1976 A monthly clinic in my outpatients at Seacroft was reserved for children with CF – the start of the Leeds CF Service.
In 1975/6, soon after neonatal CF screening started at St Mary’s, I started to follow-up the first few screened CF infants, and with the agreement of the nursing staff, we set aside a monthly Monday afternoon paediatric clinic session at Seacroft for the purpose.

The majority of the children’s medical beds in Leeds were situated at Seacroft and it was my main base where I had a major clinical commitment up to 1980 when wee movie to StJames University Hospital. I had an office and my first full time secretary, Christine Silburn, who came straight from college in 1971, worked with me until I retired in 1997 and then eventually as as senior manager of the Regional Paediatric CF Unit in Leeds until she eventually retired in 2019! A wonderful lady and awesome secretary.
In this CF clinic venture I had the enthusiastic support of Sister Moriaty the nurse in charge of Seacroft outpatients, the paediatric physiotherapist (Miss Jenkins) and the paediatric dietitian. June Wilson, was Sister on one of the paediatric wards where all my beds were situated at that time.

In the photo is the small outpatient building where first CF clinic was held. The outpatient Sister Moriarty and ward sisters also became increasingly interested and knowledgeable about CF as more children with condition came to the ward either for investigations such as sweat tests or for treatment admissions. The importance of the support and interest of staff with special expertise cannot be over emphasised.
Paediatric colleagues in the city and region gradually became aware of our new Monday afternoon monthly CF clinic and of my increasing interest in the condition. When the Professor of Paediatrics, Dick Smithells, informed all paediatricians in the Leeds Region of the special clinics that were available in his department at the General Infirmary, I took the opportunity to inform our paediatric colleagues in the Region of my CF clinic at Seacroft! By the late Seventies I was also running special clinic sessions designated for gastroenterology, asthma, diabetes and urinary tract infections (this with Professor Roy Meadow and Mr. Bob Williams, a distinguished local urologist). This may seem rather excessive but I was now an established and busy Leeds consultant and receiving many referrals; it seemed reasonable to assemble children with certain chronic problems into special clinics where one could concentrate the mind on their problems and they could be seen and discussed with various expert colleagues.
So much of the work on this first early paper on CF screening was organised by Dr Robert Evans consultant chemical pathologist at St James’s, described one of the important early foundations of the Leeds CF Service which steadily expanded so that by 2019 it was caring for 236 children at the Leeds General Infirmary and 398 adults at a modern specially adapted inpatient unit at St James’s University Hospital and with a purpose built outpatients at Seacroft Hospital.
Congden PJ. Bruce G. Rothburn MM. Clarke PC. Littlewood JM. Kelleher J. Losowsky MS. Vitamin status in treated patients with cystic fibrosis. [ Journal Article] Archives of Disease in Childhood. 56(9):708-14, 1981 Sep. https://pubmed.ncbi.nlm.nih.gov/7294874/
(Please note Peter Congdon’s name spelt incorrectly in original)
A word about some of the authors.
Dr Michael Rothburn was eventually a medical microbiologist in Liverpool until he retired in 2015 to Hale in Cheshire. He is still available as an expert witness
Dr Paddy Clarke, as he was known, moved to Harrogate from Belfast in 1969 as consultant paediatrician and also had sessions in Leeds at Seacroft. He was a great character, a good doctor very popular with the families. His wife Pat still lives in Harrogate where she is a gardening expert and has eight grandchildren
This was our first major CF study, carried out in the two years before we moved from Seacroft to St James’s in 1980. We performed an evaluation of the present nutritional state of our CF children patients (there were no adults at that time) with particular reference to the vitamin status, a particular interest of Dr. Jerry Kelleher’s, the senior biochemist in Professor Monty Losowsky’s department at St James’s. Also, we enlisted a few additional children with CF attending the clinics of interested (friendly!) paediatric colleagues, such as Dr Paddy Clarke of Harrogate and Dr Michael Buchanan in Leeds, to reach a total of 36 children with CF and 21 age-matched non-CF children as controls. When the children attended the monthly CF clinic at Seacroft I would take a venous blood specimen and Jerry Kelleher’s wife would deliver it to his laboratory at St James’s some 2 miles down the road.
The parents of the CF children were very enthusiastic; they were obviously relieved that at long last some research was underway. They were very supportive as we always found them to be subsequently with all our research studies. I was very pleased and grateful to be cooperating with such a very professional team of experts at St James’s who were more experienced than myself or any of our team in research of this type. The study was supported by a grant from the West Riding Medical Research Trust obtained by Professor Monty Losowsky.
Continuing cooperation with members of Monty Losowsky’s Department of Medicine at St James’s, particularly Dr Jerry Kelleher and his technician Mr Mike Walters, over nearly 20 years on numerous subsequent combined projects was undoubtedly one of the major factors responsible for the success and growth of our Leeds CF service.



The summary of this first study published in 1981
The late Dr. Peter Congdon was our Senior Registrar at the time. He played a major part in coordinating this and other studies during his time with me.
 The water-soluble (B1, B2, B6, C, folic acid)and fat-soluble vitamin (A, carotene, E, and D) status of the 36 patients with cystic fibrosis was assessed and compared with a control group of 21 age-matched non-CF children. Twenty-seven of the patients were receiving vitamin supplements (except for folic acid and vitamin E) at the time of investigation. Vitamin B1, B2, and B6 status was adequate in all patients (figure), and there was little evidence of folic acid deficiency. Vitamin C stores might not have been adequate in some of these patients, despite daily supplements with 50 mg of the vitamin.
The water-soluble (B1, B2, B6, C, folic acid)and fat-soluble vitamin (A, carotene, E, and D) status of the 36 patients with cystic fibrosis was assessed and compared with a control group of 21 age-matched non-CF children. Twenty-seven of the patients were receiving vitamin supplements (except for folic acid and vitamin E) at the time of investigation. Vitamin B1, B2, and B6 status was adequate in all patients (figure), and there was little evidence of folic acid deficiency. Vitamin C stores might not have been adequate in some of these patients, despite daily supplements with 50 mg of the vitamin.

Steatorrhoea, often severe, was present in most of the children. Only 4 of 24 patients had a normal fecal fat (<17.6mmol or <5g per 24hrs) despite pancreatic supplements. Steatorrhoea was often severe: 10 patients had values greater than 100mmol (28.4g)/24hrs and 17 had values greater than 50mmol (14.2g)/24Hrs.Dr. Kelleher’s laboratory with Mr. Mike Walters performing fecal fat estimations (available in very few hospitals at the time), plasma vitamin levels and subsequently many other investigations not available in most hospitals, was central to much of our research in the years that followed

Serum carotene and vitamin E concentrations were low in over 90% of patients and were related to the severity of steatorrhoea. Vitamin A was low in over 40% of the patients despite daily vitamin supplements of 4000 IU and correlated with the serum retinol-binding protein level. Serum 25-OH cholecalciferol was low in some patients whether or not they were receiving 400 IU vitamin D and correlated with the serum retinol-binding protein level. Except for serum vitamin A, which was lowest in patients with the poorest clinical grading, the other vitamins were not influenced by the clinical grade of the patients.

A subsequent short-term supplementation trial (figures above) with water-miscible preparations of vitamin A (4000IU) and E (50mg) in 14 patients, the serum levels vitamin E and 4000 IU vitamin A responded well to 2 weeks of treatment
So fat-soluble vitamin status and intestinal malabsorption were two important areas where treatment quite obviously could be much improved in many patients.
Certainly as far as these two vitamins A and E and the pancreatic enzymes were concerned, it was now obvious that in the late Seventies many of our CF children were receiving seriously suboptimal treatment. I returned to Leeds from Toronto determined to improve the care of our CF children. It was at this stage that I decided to see what other areas of management could be improved. This was the start of our “Comprehensive CF Assessments” along the lines that Douglas Crozier had recommended in 1975 i.e. “to perform a complete assessment of the patient” and then make “continuing attempts to obtain normal bodily function and maintain it”. We were on our way to developing the Leeds Regional Cystic Fibrosis Centre!
May 1980. I attend the North American Cystic Fibrosis Conference in Toronto

The importance of this 1980 North American Cystic Fibrosis Conference as a stimulus at the start of our Leeds unit warrants a little more detail. The conference was held at the impressive Royal York Hotel in Toronto. The hotel was built by the Canadian Pacific Railway company. Opened on 11 June 1929, the building has undergone several extensive renovations since it first opened, with its first major renovation in 1972. An underground walkway linking the hotel with the Royal Bank Plaza and Union Station form part of the Toronto’s underground city system – a necessary facility in the very cold winters.
I attended this conference and, with some trepidation and gave an oral presentation of the results of our detailed nutritional assessment on our few CF children. I also presented a poster reporting our neonatal screening results from St Mary’s Hospital in Leeds. Both these studies were later published.
– Congdon PJ, Bruce G, Rothburn MM, Clarke PCN, Littlewood JM, Kelleher J, Losowsky MS. Vitamin status in treated patients with cystic fibrosis. Arch Dis Child 1981; 56:708-714.https://pubmed.ncbi.nlm.nih.gov/7294874/
– Evans RT, Little AJ, Steel AE, Littlewood JM. Satisfactory screening for cystic fibrosis with the BM meconium procedure. J Clin Path 1981; 34:911-913.
For my part, although by now a very busy and increasingly experienced general consultant paediatrician for some 12 years, as far as CF was concerned, I definitely felt a relative rookie at this conference. Attendance at this excellent conference was the start a very steep learning curve for me and subsequently my immediate colleagues at St James’s. Undoubtedly, attendance at this Toronto conference had a major influence on my approach to treating children with CF and represented a definite turning point in my professional life in that CF would eventually become one of my main interests.

Also at this 1980 meeting I had the opportunity to visit the CF Centre at the Sick Children’s Hospital in Toronto and talk with the CF Centre Director, Dr Henry Levison. At the main meeting it was announced that anyone wishing to visit the respiratory unit would be welcome. I recall there were only four of us took up the offer, Dr Archie Norman, Mrs Norman, myself and another doctor. Henry saw us amid a vast collection of complex machines which made our Vitalograph, a small table top machine for testing respiratory function we used in Leeds, seem rather inadequate!


Henry was a definite leader and involved in many of the earlier advances in paedaitric care in Toronto
On the lighter side, during this first visit I enquired if Henry checked the children with CF and asthma for allergies. Henry appeared horrified and exclaimed “Oh no! You’re not a bloody allergist are you?. Although I denied this firmly at the time, whenever we met subsequently at meetings it was “Here’s the allergist from Leeds!” I visited him again in Toronto some 6 years later and was equally overwhelmed and impressed. He was also a recognised art expert.

Dr Douglas Crozier (1940-2014) started the Toronto CF clinic in 1958, the second in Canada after Dr Alan Ross had opened the first at Montreal Children’s Hospital. In 1974 Crozier wrote “success of treatment will depend on a complete assessment of the patient and then continuing attempts to obtain normal bodily function and maintain it”. He described how he advised his patients to abandon the traditional low fat diet and used very high doses of pancreatic enzymes (up to 100 Cotazym pancreatic enzyme capsules per day!). Crozier believed that “to deprive a child with cystic fibrosis, who usually has very little subcutaneous fat, of this important nutrient seems ridiculous” The superior nutritional state of the Toronto patients from this approach in the Seventies is believed to be the main reason for their significantly better survival compared to those in the USA. As far back as 1973, 428 people with CF were attending the Toronto clinic of whom 92 (21.4%) were 16 years or older, this was quite remarkable for that time e.g. when there were few adults in the UK.
I was told by a paediatrician who had worked with Douglas Crozier for a time that he was very thorough always attending the patients’ postmortems and analysing how his treatment could be improved in the management of his patients through their lives and correlating these facts with the post-mortem findings. Although I was never fortunate enough to meet Crozier I was profoundly impressed by his landmark paper from Toronto regarding the approach to management of people with cystic fibrosis.
1974 Crozier DN. Cystic fibrosis: a not so fatal disease. Pediatr Clin North Am 1974; 21:935-948. https://pubmed.ncbi.nlm.nih.gov/4610494/
Undoubtedly, Douglas Crozier’s positive approach to the treatment of children with CF had a major influence on my subsequent approach to management of the condition.
Congdon PJ. Littlewood JM. Aggarwal RK. Shapiro H. Glucose 6-phosphate dehydrogenase deficiency and cystic fibrosis. [ Case Reports. Journal Article] Postgraduate Medical Journal. 57(669):453-4, 1981 Jul. https://pubmed.ncbi.nlm.nih.gov/7312744/
A 2.85 kg infant born to unrelated Pakistani parents is described. He was diagnosed as having cystic fibrosis by neonatal screening and started on standard treatment with pancreatic replacements (Cotazym), oral cloxacillin and physiotherapy was started.
At 5 weeks the infant was noted to be very pale but not jaundiced. Investigations revealed he was severely anaemic with a haemoglobin was only 5.6g/dl, WBC 8’3xlO0/l; platelets 355×109/l reticulocytes 17%; serum iron 15 mmol/l (normal 14-31 mmol/l) and serum folate 10.5 ug/l (normal 2-5-20 ug/l), vitamin E 420 mg/dl (normal 520-1140 mg/dl). The dye decolorisation screening test for G-6PD-deficiency was positive and subsequently no G-6PD activity was detected.
He was transfused with fresh blood and subsequent progress was satisfactory with no further evidence of haemolysis. He continued to thrive on pancreatic supplements but no antibiotics. There has been no further evidence of haemolysis, and haemoglobin levels have remained between 9.5-11.5 g/dl. The dye decolorization time in the mother was >7hr, noG-6PD activity was detected in her red cells. The father has not been investigated.
Both these conditions in the same individual had not previously been reported. Glucose 6 phosphate dehydrogenase (G6PD) deficiency is a hereditary condition in which red blood cells break down (haemolysis) when the body is exposed to certain foods, drugs, infections or stress. It occurs when a person is missing or has low levels of the enzyme glucose-6-phosphate dehydrogenase.
Apparently about 400 million people have the condition globally. It is particularly common in certain parts of Africa, Asia, the Mediterranean, and the Middle East. Males are affected more often than females. In 2015 it is believed to have resulted in 33,000 deaths (Wikipedia).
Congdon PJ. Fiddler GI. Littlewood JM. Scott O. Coeliac disease associated with congenital heart disease. [ Journal Article] Archives of Disease in Childhood. 57(1):78-9, 1982 Jan. https://pubmed.ncbi.nlm.nih.gov/7065700/
There may be more than one reason for a child’s poor growth and/or symptoms. Doctors are often warned against “double pathologies” but this does not seem to apply to paediatrics. For example, an infant was referred by Dr Olive Scott, our distinguished paediatric cardiologist (1924-2007). Based on her extensive experience, there was more interference with the infant’s growth than she would expect from the severity of the cardiovascular abnormality.
As part of our investigations of this infant that Olive Scott referred, a jejunal biopsy was performed which revealed subtotal villous atrophy and confirmed coeliac disease as an additional factor affecting growth. Following this experience we saw a further five children with congenital heart disease referred by the paediatric cardiologists in whom poor growth was found to be due to small-bowel villous atrophy; none was in heart failure and only one was severely cyanosed. Growth improved in all 6 on a gluten-free diet.
We suggested that gluten enteropathy may be more common than is realised in children with congenital heart disease, and jejunal biopsy should be undertaken early in any patient with poor growth and no heart failure to exclude the coexistence of the condition. However, in recent times, of course, appropriate blood antibody tests would be the first step. The European Society for Paediatric Gastroenterology, Hepatology, and Nutrition (ESPGHAN) 2019 guidelines for the diagnosis of celiac disease state that if an anti-tissue transglutaminase antibody test result is greater than 10 times the manufacturer’s upper limit of normal range, then endoscopy can be avoided in favour of confirmatory endomysial antibody (EMA) testing.

Dr Olive Scott trained in Liverpool and moved to Leeds when her husband James was appointed Professor of Obstetrics. After working at the General Infirmary she was appointed by Killingbeck Hospital as consultant paediatric cardiologist – the first person in Great Britain to hold such a position. She was the first person in the UK to perform balloon atrial septostomy and developed non-invasive diagnosis through cardiology. A national expert and a very pleasant colleague.
Dr Garrick Fiddler, the other c-author, after a number cardiology appointments dating from 1974 last published in 1990 when at the Glaxo Group Research Ltd Greenford.
Brown RC. Chalmers DM. Rowe VL. Kelleher J. Littlewood JM. Losowsky MS. Comparison of the diagnostic value of serum pancreatic isoamylase and immunoreactive trypsin measurement in patients with cystic fibrosis. [ Journal Article] J Clin Pathol 35(5):547-9, 1982 May. https://pubmed.ncbi.nlm.nih.gov/7085899/
This study was coordinated by two of Professor Losowsky’ lecturers in medicine at St James’s – Richard Brown and Douglas Chalmers. Richard Brown became a consultant gastroenterologist in Glasgow and Doug Chalmers remained in Leeds as consultant gastroenterologist at Leeds General Infirmary – his last publication being in 2004.
Serum immunoreactive trypsin (IRT) and serum pancreatic iso-amylase (PIA) activities were measured using commercially available kits in 37 cystic fibrosis patients and 46 hospital controls of similar age range. Immunoreactive trypsin was more often abnormal than PIA (26/37 v 18/37 abnormal respectively); IRT will be particularly useful as an additional diagnostic test in older children, in whom interpretation of the sweat test may be difficult, as 14/15 CF patients aged over 10 years had abnormal IRT results. Less than half of our patients who were aged between one and nine years had abnormal IRT activity, limiting the value of the test, though a low activity would still support the diagnosis of CF. Comparison with faecal fat estimations in 31 patients suggests that neither IRT nor PIA can be used as a non-invasive test of pancreatic function in order to identify those few CF patients who do not require pancreatic enzyme supplements.
So we found the IRT to be of limited use in CF patients in contrast to its great value in neonatal screening.
Minford AM. MacDonald A. Littlewood JM. Food intolerance and food allergy in children: a review of 68 cases. [ Case Reports. Journal Article] Arch Dis Child 57(10):742-7, 1982 https://pubmed.ncbi.nlm.nih.gov/7138062/
Over a 10-year period the diagnosis of food allergy/intolerance was considered likely in 121 children referred to my clinic. Eventually 68 were considered to have definite food intolerance and food allergy. Forty eight (70%) presented with gastrointestinal symptoms and signs.(vomiting, diarrhoea, colic, abdominal pain, failure to thrive), 16 (24%) children had skin manifestations (eczema, urticaria, angioneurotic oedema, other rashes), and 4 (6%) children with wheeze. Twenty-one children had failed to thrive before diagnosis. A single food (most commonly cows milk) was concerned in 28 (41%) cases. Forty (59%) children had multiple food intolerance or allergy; eggs, cows milk, and wheat were the most common. Diagnosis was based on observing the effect of food withdrawal and of subsequent re-challenge. In many children food withdrawal will mean the use of an elimination diet which requires careful supervision by a dietitian experienced in this area. Laboratory investigations were often unhelpful in suggesting or confirming the diagnosis. Certainly the involvement of an experienced dietitian is absolutely essential.

I was fortunate that we had Anita MacDonald, an outstanding paediatric dietitian working at St James’s. Anita was an important co-author of a number of publications both on food intolerance and cystic fibrosis. She eventually moved to Birmingham Children’s Hospital as Chief Dietitian where she eventually became a professor and later was awarded an OBE. Some dietitian! We were very fortunate to have her at St James’s during the Eighties as our senior paediatric dietitian.
Cows’ milk protein intolerance is a common cause of three month colic in babies. Severe infantile colic driving the unfortunate parents to distraction was not an uncommon problem referred to me. The mother had usually been reassured by the family doctor (and sometimes by a consultant paediatrician!) that it would settle and the baby was healthy. A 48 hour trial on the hydrolysed protein feed Nutramigen usually settled the colic completely. The usual response from the relieved parents was “Why didn’t my doctor put him on this feed?”


The relationship of severe 3-month colic and other gastrointestinal disorders to cows milk intolerance was a striking and not uncommon problem in my experience. Failure to recognise the association between gastrointestinal signs and cows milk intolerance at times resulted in unnecessary operations for gastro-oesophageal reflux before the cause of the infant’s symptoms was identified as in the infant illustrated here.
This infant who whose gastro-oesophageal reflux failed to respond to both medical treatment and then an unnecessary fundoplication operation was cured on a cows’ milk free diet
The other variation on this problem was the breast-fed infant who is also obtaining small amounts of cows’ milk protein antigens from the breast milk. Stopping breast feeding and starting Nutramigen would usually cure the problem. Putting the mother on a cows’ milk free diet was another treatment option but always seemed rather involved for the family who were already stressed by their infant’s problems. I must confess I usually opted for Nutramigen.
The other really interesting aspect of this association of 3-month colic and cow’s milk intolerance was the refusal of many otherwise sensible consultant paediatricians (including one friendly Professor of Paediatrics!) to doubt the importance of the cow’s milk in these unfortunate infants and who fail to give trial to a cows’ milk free diet. In all seriousness, I felt very strongly about failure to diagnose these infants as, at a time for when the parents should have been enjoying the first months of the their new baby, they were made to feel inadequate and a failure in caring for their infant – sadly often by their family doctor or health workers. At least one of my seven grandchildren did well on Nutramigen!
Minford A M B, MAcdonald A, Littlewood J M. Food intolerance and food allergy in children:a review of 68 cases. Arch Dis Child 1982; 57:742-747.Free https://pubmed.ncbi.nlm.nih.gov/7138062/
Littlewood JM, Macdonald A. Clinical aspects of food allergy and intolerance. In Heatley RV., Losowsky MS, Kelleher J. (eds.) Clinical Nutrition in Gastroenterology 1986 Â pp202-33. Â Churchill Livingstone.
Littlewood JM, Macdonald A. Food Intolerance: Our Practice. Nutrition and Health 1987: 5 (3/4):119-135. A B Academic Publishers.
Littlewood JM. Allergy of the gastrointestinal tract. In Recognition and Management of Food Allergy in Children. Franklin A J (ed). 1988 pp 21-29. The Parthenon Publishing Group.
Adrian Minford subsequently became a consultant paediatrician in Bradford where he had a successful career. He wrote a book in 1998 “Illustrative Signs in Clinical Paediatrics”.
Yohannan MD. Terry HJ. Littlewood JM. Long term phototherapy in Crigler-Najjar syndrome. [ Case Reports. Journal Article] Archives of Disease in Childhood. 58(6):460-2, 1983 Jun. https://pubmed.ncbi.nlm.nih.gov/6859942/
This article is discussed after the first report of this child by Bill Arrowsmith in 1975 above
Ryatt KS. Cotterill JA. Littlewood JM. Generalized pruritus in a baby as a presenting feature of the arteriohepatic dysplasia syndrome. [ Case Reports. Journal Article] Clinical & Experimental Dermatology. 8(6):657-61, 1983 Nov. https://pubmed.ncbi.nlm.nih.gov/6661830
I was asked to see this child by the paediatrician in Harrogate Hospital. A 2 year old boy had been adopted at 9 days and had scratched his skin intensely since that time. He did have transient neonatal jaundice. His general milestones and growth had been normal. He had extensive skin excoriations without evidence of eczema. His appearance was slightly unusual in that his forehead was prominent, his nose rather straight, his chin was small pointed and the eyes were set deeply and somewhat widely apart (figure of another child with syndrome).

There was an obvious systolic heart murmur audible over the whole chest. The liver was palpable one finger below the costal margin. Extensive investigations were normal apart from liver function tests – alkaline phosphate 71.4 (N 0-20 KA units), ALT 82 iu/l (N 0-40) and the plasma 25hydroxy vitamin D, 1,2ng/mi was very low in the rickets range.

I performed a needle biopsy of the liver which was initially considered normal but Dr Alex Mowat a colleague and national liver expert at King’s College Hospital, London considered it to be compatible with the diagnosis of arteriohepatic dysplasia (Alagille syndrome) showing intrahepatic bile duct hypoplasia and bile thrombi. Mild pulmonary stenosis was confirmed by Dr Gordon Williams our paediatric cardiologist.

We tried various treatments for the pruritus. Three different regimens of antihistamines, topical corticosteroids and a cows’ milk free diet and all failed. However, cholestyramine, reducing cholesterol and bile acid levels, produced a dramatic reduction in pruritus but this returned if ever the dose was less than 2.5g/day. Vitamins D and other fat soluble vitamins were also given.
In our article we reviewed previous patients described since 1949. There is now (2021) an extensive literature dealing with the condition including 91 liver transplantations eventually required in 20% to 50% of cases.
Dr John A Cotterill was a consultant dermatologist in Leeds born in 1940. He seems to have been very active in the dermatology world. He seems to be critical of the advertising industry. At the 1987 World Congress of Dermatology his contribution on Body Image and the Advertising Industry he states that “Western woman in encouraged therefore by advertising industry to be entirely infantile in her ideas about her skin and in her skin care” – a concept that he enlarges upon. He has 116 references listed on PubMed from 1970 to 2002 – the most recent on damage limitation in cosmetic dermatology!
Dr Gordon Williams was a consultant at the Yorkshire Regional Cardiothoracic Centre where he shared much of the paediatric cardiology with Dr Olive Scott. He eventually moved to the York Teaching Hospitals NHS Foundation Trust and appeared to do more medical legal work.
Alex Mowatt (1935-1995) was a Scottish paediatric hepatologist. He established the paediatric hepatology unit at King’s College Hospital, London, which became a referral centre for children across Britain with liver diseases. He was appointed professor of paediatric hepatology in 1990. He was a very pleasant unassuming man whom I knew reasonably well. Hence I sent him this patient’s liver biopsy for his opinion.
The syndrome is now called Alagille’s syndrome and it is one of the most frequent causes of a paucity of intra-hepatic bile ducts. Apparently prognosis is good for survival but patients are likely to have pruritus, xanthomas, elevated cholesterol levels and neurological complications of vitamin E deficiency if untreated.
Bennett MJ. Littlewood JM. MacDonald A. Pollitt RJ. Thompson J. A case of beta-ketothiolase deficiency. [ Case Reports. Journal Article] Journal of Inherited Metabolic Disease. 6(4):157, 1983. https://pubmed.ncbi.nlm.nih.gov/6422156/
The patient, A.K., is a female, the first child of unrelated Caucasian parents. The family also includes a younger unaffected male sibling. At 5 months A.K. was admitted to St. James Hospital, Leeds, following a history of 3 days vomiting and several hours rapid grunting respiration. She appeared extremely ill, with pyrexia, dehydration and acidotic breathing (60/min). She had vomited altered blood, and subsequently passed a melaenic stool. There was a marked metabolic acidosis: plasma sodium 140mmol/1, potassium4.7mmol/1, total COz 7retool/l, urea 9.4mmol/1, chloride 105mmol/1, and a raised plasma aspartate aminotransferase activity of 145 units (reference range 3-35 units). There was a marked improvement upon rehydration and correction of the acidosis.
At 17 months a similar episode occurred coincidental with acute gastroenteritis. Again she was severely acidotic with a plasma total CO2 of 3mmol/1 and a blood pH of 6.99. On this occasion there was no pyrexia and no melaena. There was a rapid response to intravenous fluid and alkali but the severity of the acidosis suggested an underlying metabolic abnormality which was then pursued, diagnosis being made at 24 months.
Analysis of urinary organic acids by gas chromatography-mass spectrometry of trimethylsilyl derivatives after discontinuous solvent extraction revealed a large excess of 2-methyl-3-hydroxybutyric acid (0.98mmol/mmol creatinine) and tiglylglycine.
These are characteristic findings in 2-methyIacetoacetyl- CoA thiolase (13-ketothiolase) deficiency (McKusick 20375; Chalmers and Lawson, 1981). A protein load led to a further increase in the excretion of 2-methyl-3- hydroxybutyric acid (4.4mmol/mmol creatinine) and tiglylglycine, and also to the excretion of 2-methylaceto- acetic, 2-methyl-3-ketobutyric, (E)-2-methylglutaconic, and 2,3-dimethyl-3-hydroxyglutaric acids. Enzyme assay of cultured skin fibroblasts performed by Dr B. Middleton, Nottingham University Medical School, revealed no detectable activity of 2-methylacetoacetyl- CoA thiolase, and normal activity of other thiolase enzymes. Nine previous cases of this condition have been described although only two others have been confirmed by direct enzyme analysis (Schutgens et al., 1982).
A dietary history revealed a very high intake of 68 g protein day-1 (body weight 10kg). This has been reduced to2g kg A.K.is generally in good health. However an acidotic attack associated with increased excretion of 2-methyl-3-hydroxybutyrate has occurred since dietary restriction. This coincided with gastroenteritis which affected other members of the family. When asymptomatic the excretion of 2-methyl-3- hydroxybutyrate appears to be consistently in the range 0,4-0.5 mmol/mmol creatinine.
Beta-ketothiolase deficiency is an inherited disorder in which the body cannot effectively process a protein building block (amino acid) called isoleucine. This disorder also impairs the body’s ability to process ketones, which are molecules produced during the breakdown of fats.
Apparently signs and symptoms typically appear between the ages of 6 and 24 months. Affected children experience intermittent episodes of ketoacidosis, characterized by vomiting, dehydration, difficulty breathing, extreme tiredness (lethargy), and occasionally, seizures. In severe cases, these episodes can lead to coma. Metabolic stroke is another finding that has been increasingly reported in children with this condition. Ketoacidotic attacks are frequently triggered by infections, periods without food (fasting), or increased intake of protein-rich foods.

Beta keto-thiolase deficiency is inherited in an autosomal recessive fashion and is caused by pathogenic variants (mutations) in the ACAT1 gene. Treatment involves managing acute crises with intravenous (IV) fluids, glucose, and electrolytes along with bicarbonate. Long-term management involves eating frequently, following a reduced-protein diet, avoidance of high-fat foods, and, in some cases, carnitine supplementation.
Dr Michael Bennett at the time of this report was Clinical Chemist at Sheffield Children’s Hospital; he eventually became Professor of Pathology & Laboratory Medicine at the University of Pennsylvania Perelman School of Medicine, USA.
Conway SP. Gillies DR. Littlewood JM. Vitamin B12 neuropathy in a 6 year old. [ Case Reports. Journal Article] Archives of Disease in Childhood. 59(6):575-6, 1984 Jun. https://pubmed.ncbi.nlm.nih.gov/6742880/ FREE
A Leeds family doctor asked if I would see a child whose parents had recently brought their 6 year old daughter from Bangladesh in view of a serious progressive illness. She had been well to the age of 3 years when her tongue became red and painful; she suffered unexplained episodes of severe anaemia despite repeated courses of iron and folic acid and there was a gradual onset of muscular weakness. From 5 years she developed a coarse tremor of the limbs and face.
On examination the child was weak and wasted (weight < 3rd and height 10th centile). She had difficulty swallowing, had slurred speech and saliva drooled from the side of her mouth. Her tongue was red, wasted and showed fasciculation.
Initial assessment suggested a brain stem tumour but computed tomography showed unexplained mild hydrocephalus and a general decrease in size of other parts of the brainstem. Extensive metabolic investigations were also normal. However a second blood sample taken during a febrile episode was abnormal showing severe pancytopenia prompting bone marrow examination which was also very abnormal and megaloblastic (cells larger than normal). The serum B12 was only 55 ng/l (low normal range 110ng/l. Further tests confirmed gross B12 deficiency which we showed was due to defective absorption.
Treatment with vitamin B12 1 mg daily brought about an almost miraculous improvement. There was rapid response in the anaemia and bone marrow. Growth and weight accelerated (Ht to 25th, Wt to more than 25th centile) over 4 months and she became an active child, able to run unaided. The only residual abnormalities were some loss of peripheral sensation and absent tendon reflexes with slight reduction in power.
We concluded, by exclusion, that the condition was due to an inherited failure to absorb vitamin B12 from the small intestine. This was really a remarkable case. The parents and referring doctor were obviously delighted. It was a good example of how many experts in a massive hospital, such as St James’s, working together solved a very serious and difficult problem.

Dr Steve Conway was my general paediatric registrar at the time of this report. He subsequently went on to form the Leeds Adult Cystic Fibrosis Unit and finally become Director of both the Paediatric and Adult Leeds Regional CF Units at Seacroft and St James’s and also became a leading international authority on cystic fibrosis. Steve and his wife Ella have remained our good friends over the years.
Howdle PD. Littlewood JM. Firth J. Losowsky MS. Routine colonoscopy service. [ Journal Article] Archives of Disease in Childhood. 59(8):790-3, 1984 Aug. FREE https://pubmed.ncbi.nlm.nih.gov/6476884/


Dr Peter Howdle, then a Senior Lecturer in Monty Losowsky’s department, was a quite brilliant colonoscopist. Apparently in 1977 Monty asked Peter to develop a colonoscopy service at St James’s. It was predicted, by experienced trainers, that it would take about 50 colonoscopies before a colonoscopist could reliably reach the caecum at each examination. The hand-written record Peter kept shows he managed this in only 35 examinations (from A History of Specialities in Leeds Teaching Hospitals NHS Trust 1767-2018).
So Peter was an expert colonoscopist and eventually with him as main expert we developed a paediatric colonoscopy service working in our paediatric Day Unit.

The arrangement and procedures were managed very efficiently by Nurse Jeanette Firth whose sole responsibility was running the day Unit. She had been fully trained in the adult endoscopy unit. Children were admitted to the adjacent ward two days or the day before the procedure for bowel preparation and the procedure performed under sedation with pethidine and diazepam. Occasional general anaesthetic was used for example for polypectomy (as in the figure). The details of the procedure are in the full version of the paper which is available via the PubMed. Forty children aged between 7 months and 16 years underwent 50 colonoscopies between march 1978 and October 1983. Inflammatory bowel disease (18), colonic polyps (7), rectal bleeding (10), family polyposis were the main diagnoses.
The paediatric colonoscope was used by the paediatrician (JML) for sigmoidoscopies with the children only attending for the day – a valuable and relative technically easy non-stressful procedure.
Littlewood JM, Kelleher J, Losowsky MS, Page R, Crollick AJ, Miller MG, Conway SP, Firth J, MacDonald A, Henry F. Comprehensive clinical and laboratory assessment in cystic fibrosis. From Cystic Fibrosis:Horizons. Ed.Lawson D, John Wiley and Sons, Chichester 1984:266 Published only in conference proceedings.
 This was in the Proceedings of the 9th International Cystic Fibrosis Congress , Brighton, England June 9th – 15th, 1984
This was in the Proceedings of the 9th International Cystic Fibrosis Congress , Brighton, England June 9th – 15th, 1984








This was the first publication of significant number of our Comprehensive Assessments the provision of which had already resulted in a rapid growth of our St James’s CF clinic. We started Comprehensive Assessments for our own CF patients in May 1980 when I returned from the North American CF Meeting in Toronto. They proved to be so useful for the management of our own patients that the following year I offered the service to consultant paediatricians in the Yorkshire Region. They also found them useful and in 1983 with their support the Yorkshire Regional Health Authority recognised us as providing a regional CF service at St James’s.
Here we describe 103 CF patients from the Yorkshire Region who had a comprehensive clinical, laboratory, radiological, dietetic and physiotherapy assessments. In a further five patients previously considered to have CF by their referring consultants the diagnosis was not confirmed and an alternative diagnosis established
They were 55 males and 48 females. The average age was 8.2 years. The the mean Shwachman clinical score was 74 (ranging from 100 to 30) and the mean Chrispin-Norman x-ray score was 9 at the time of the initial assessment 57 patients were attending our CF clinic at 46 were referred from the clinics of 20 other paediatricians and chest physicians in the region.
Delays in diagnosis were common (20% > a year before diagnosis; 27% > six months and 40% > three months) Ten families had no knowledge of the national charity, the Cystic fibrosis Trust. Twenty two siblings had not had a sweat test.
Detailed clinical laboratory and x-ray evaluation is revealed evidence of active chest infection and 34 patients, 34 had inflammatory infiltrations on the chest x-ray, 24 had an absolute neutrophil count greater than 7500 per cubic millimetre, nine patients were going Staph. aureus or H Influenzaea or both and not on an appropriate antibiotic, 23 patients had an abnormal ESR of more than 25, 27 had a total of white blood count of more than 12,000 .These results frequently resulted in more aggressive management of the chest infection.
Significant intestinal malabsorption persisted in 78 treated patients and evidence of gastrointestinal involvement was invariable. Subnormal fat soluble vitamin levels were present – vitamin A 68%, vitamin E 88%. Mineral levels were normal except for iron which was low in 43%
Despite the usually recommended daily energy intake of 120% IDI only 21 patients out of 86 achieved this in practice. In many patients total energy intake was compromised by their adhering to the traditional low fat diet.
In the majority of patients there were multiple areas where changes in therapy were indicated. The expectations of doctors and parents were often low by modern standards. Regular comprehensive assessment by a team experienced in cystic fibrosis will significantly improve the management of quality of life of CF patients in the UK. The “tertiary referral service” we have described appears to be accepted and used by the majority of consultant paediatricians in our region and eventually in the UK
The co-authors represented the team who had undertaken the assessments. Jerry Kelleher (biochemist), Avril Crollick (paediatric clinical assistant), Mike Miller (cystic fibrosis research fellow), Steve Conway (paediatric registrar), Jeanette Firth (coordinating Day Unit Nurse), Anita MacDonald (senior paediatric dietitian) and Fiona Henry (paediatric physiotherapist. Monty Losowsky (Professor of Medicine) although not hands on practically involved was a major source of encouragement and advice. Richard Page (respiratory physician) had a few adult patients. Not mentioned is the secretarial support and organisation from Christine Silburn and paediatric secretaries and also the increasingly complex data management by Ann Littlewood SRN.
Wilson-Sharp RC. Irving HC. Brown RC. Chalmers DM. Littlewood JM. Ultrasonography of the pancreas, liver, and biliary system in cystic fibrosis. [ Journal Article] Archives of Disease in Childhood. 59(10):923-6, 1984 Oct. https://pubmed.ncbi.nlm.nih.gov/6388511/

Dr R C Wilson-Sharp was a radiology registrar at St James’s and Henry Irving a consultant radiologist there. As was my policy, I would try to interest any likely colleagues to apply their skills to the investigation and/or treatment of our increasing number of patients with cystic fibrosis. There were few studies of the liver involvement in people with CF at that time in 1984.
Dr Wilson-Sharp performed abdominal ultrasound imaging in 50 children – 39 with CF and 11 with other forms of respiratory problems. The pancreas was abnormal in 75% of CF patients under 5 years, in 95% over 5 years and in all who had malabsorption. Liver parenchyma was abnormal in 23%, of the gall bladder in 24% and splenomegaly in 8% of the CF patients. The relatively poor correlation between the biochemical liver function tests and ultrasonographic liver involvement is due to the focal nature of the the liver disease.


Gray RG. Lowther GW. Littlewood JM. Middleton B. Bennett MJ. A case of 2-methylacetoacetyl CoA thiolase deficiency with coincidental chromosome abnormalities. [ Case Reports. Journal Article] Journal of Medical Genetics. 21(5):397, 1984 Oct. https://pubmed.ncbi.nlm.nih.gov/6150117/
Mitochondrial acetoacetyl-CoA thiolase (T2) deficiency is an inborn error of metabolism that affects the catabolism of isoleucine and ketone bodies. This disorder is characterized by intermittent ketoacidotic episodes. The investigation was a combined effort to solve a rare problem of an infant admitted under my care who presented at the age of 5 months with vomiting, pyrexia, dehydration and a metabolic acidosis associated with increased urinary excretion of 2—methyl-3-hydroxybutyrate, triglycine, 2-methylacetoacetate2-methyl-3-ketobutyrate, (E)-2-methylglutaconate and 2-3-dimethyl-3-hydroxybutyrate. Cultured fibroblasts revealed a total deficiency in the activity of 2-methyl-acetacetyl CoA thiolase.
The present policy in the laboratory is to karyotype all cultured fibroblasts. This revealed in her case a 47,XXX karyotype. Phytohaemag-glutinin stimulated lymphocytes from the patient exhibited a mosaic karyotype of 45,X/47,XXX(25%/75%).
At the time of this report (3years 9 months) she is well and shows none of the external stigmata of Turner’s syndrome but may well experience problems at the onset of puberty. The frequency of acidotic attacks has decreased on reduction of her dietary protein intake
A family has also been reported in which one male sib had no detectable enzyme activity while his brother had a level of activity consistent with the heterozygous state. This would suggest that this disorder is inherited in an autosomal mode and therefore that the chromosome abnormalities in our patient are unlikely to be related to the biochemical disorder.
Chromosome abnormalities have been detected in patients with inborn errors of metabolism and in some cases the biochemical and cytogenetic abnormalities have been directly linked. Our experience with this case indicates the value of karyotyping cels cultured for biochemical tests from patients with inborn errors of metabolism.
Dr RGF Gray was in the University Sub-Department of Medical Genetics, Dr W G Lowther Centre for Human Genetics, Sheffield. Dr B Middleton Department of Biochemistry University of Nottingham and JJ Bennett of the Department of Chemical Pathology, Sheffield Children’s Hospita
Miller MG. Ghoneim AT. Littlewood JM. Use of enoxacin in a patient with cystic fibrosis. [ Case Reports. Letter] Lancet. 1(8429):646, 1985 Mar 16. https://pubmed.ncbi.nlm.nih.gov/2857989/

Enoxacin is an oral antibiotic of the quinolone class antibiotic active against many bacteria including Pseudomonas aeruginosa. In 1981 a 9-year old boy with cystic fibrosis was transferred to the Regional CF Unit. Despite numerous admissions for intravenous antibiotics he continued to deteriorate with pseudomonas aeruginosa cultured from November 1982. By 1984 he was thin and breathless hardly able to walk. Varied combinations of antibiotics intravenously were ineffective.
With consent of all parties concerned he was given oral enoxacin twice daily for 7 days as his only antibiotic. The drug was supplied by Warner Lambert International. Serum levels were adequate and the drug was well tolerated. Enoxacin 400mg twice daily was given at home for 14 day courses at 9,16, 20 and 26 weeks after the first course with no adverse effects. During the second course there was subjective improvement with reduction in cough and dyspnoea and modest improvement in respiratory function.
The use of enoxacin, an early oral anti-pseudomonal antibiotic, enabled us to provided anti-pseudomonal cover at home and extend and to some extent maintain his quality of life.
This first oral anti-pseudomonal antibiotic was exciting and welcome. It was never used widely in CF but in 1987 ciprofloxacin became available and dominated the market.
Dr Mike Miller was our first CF Research Fellow at St James’s funded by the CF Trust from 1983.- the first in the UK. The presence of an experienced registrar fully committed to CF was a massive advance in care and the development of our regional CF unit. Mike was very popular with patients, parents and staff and made a major contribution during his more than 2 years stay with us. He eventually went to Huddersfield as consultant paediatrician and then as Medical Director to Martin House the children’s hospice at Boston Spa, Wetherby.
Dr A T Gonheim was the consultant bacteriologist at St James’s drung the Eighties and he and his staff were involved in many studies involving the CF patients.
Littlewood JM. Miller MG. Ghoneim AT. Ramsden CH. Nebulised colomycin for early pseudomonas colonisation in cystic fibrosis. [ Letter] Lancet. 1(8433):865, 1985 Apr 13. https://pubmed.ncbi.nlm.nih.gov/2858720/
This letter to the Lancet from our CF Unit at St James’s University Hospital was the first report of the use of nebulised colomycin to successfully eradicate early Pseudomonas infection in children with cystic fibrosis.
Although only a modest letter, this was undoubtedly the most important publication of my career!
Pseudomonas aeruginosa once it was isolated from respiratory cultures of a person with CF usually persisted; there followed a gradual increase in a chronic cough and sputum production and deterioration in respiratory function. Without wishing to overdramatise the situation – at that stage the patient had passed the “point of no return”. Thereafter progress would be steadily downhill – the rate of decline depending on the treatment the patient received. At some stage, when the symptoms worsened the child would be treated with a course of intravenous anti-Pseudomonal antibiotics as there were no oral antibiotics effective against P.aeruginosa. In the majority, these intermittent courses of IV antibiotics would continue to the end of the patient’s life. Until the later part of the Eighties this treatment necessitated a two week admission to hospital with a major disruption to family life. Home intravenous antibiotics were not introduced until the second half of the Eighties when also oral ciprofloxacin, the first oral antibiotic effective against P. aeruginosa, also became available.
Some background: Long term nebulised anti-Pseudomonal antibiotics, such as gentamicin or tobramycin, were by no means accepted as a safe, effective treatment in the Eighties and tended to be used when the respiratory function was deteriorating rather than to eradicate early infection. However in 1981 there was a seminal publication by Dr Margaret Hodson and her colleagues at the Royal Brompton Adult CF clinic in London on the use of nebulised antibiotics in patients with chronic Pseudomonas infection.
Hodson ME et al. Aerosol carbenicillin and gentamicin treatment of Pseudomonas aeruginosa in patients with cystic fibrosis. Lancet 1981; i: 1137-1139.https://pubmed.ncbi.nlm.nih.gov/6118579/

Most adults with CF had chronic Pseudomonas infection with frequent exacerbation of their chest infection requiring admission to hospital for intravenous antibiotics. Margaret Hodson’s use of nebulised antibiotics significantly reduced the frequency of these exacerbations. Respiratory function recordings in a patient receiving nebulised antibiotics or placebo showing stabilisation with treatment. This 1981 paper of Margaret’s had a major influence on treatment in the UK although even though there had been many earlier concerns about nebulised antibiotics increasing the incidence of bacterial resistance.
Although nebulised penicillin had been used in the Forties when S. aureus was the main pathogen (di Sant’Agnese, et al, 1946) it was undoubtedly this paper from the Brompton, that revived the interest in nebulised antibiotics for patients with chronic Pseudomonas infection who were having frequent exacerbations. Although numbers in the trial were small the nebulised anti-Pseudomonal antibiotics, gentamicin and carbenicillin, one did not need a trial to see that obviously stabilised the condition of some patients with frequently relapsing chronic P, aeruginosa infection who were requiring increasingly frequent courses of inpatient IV antibiotics.
So as chronic relapsing Pseudomonas infection was a major and increasing problem even in the children, I decided to examine the possibility of treating the early P. aeruginosa infections, before the organism became established. However, I was reluctant to use one of the aminoglycosides (gentamicin or tobramycin) in our young patients as eventually these antibiotics would be needed for intravenous antibiotic treatment.
I found some old references reporting the successful use of nebulised colomycin in non-CF adult patients with pneumonia due to P. aeruginosa
– Halliday NP. Pseudomonas infection of the respiratory tract treated with colistin sulphomethate sodium (colomycin). Clin Trials J 1967 August 771-775.
– Herrell WE. Aerosolized colistimethate in the treatment of pulmonary infections. Clin Med September 1968, 18-19. Rose HD et al. Evaluation of sodium colistimethate aerosol in gram-negative infections of the respiratory tract. J Clin Pharm 1970; 19:274-281
I discussed the possibility of using nebulised colomycin in children with CF with the UK manufacturers (Pharmax at the time) from February 1981. From the information they provided it seemed reasonable to try a small dose of nebulised colomycin twice daily in an attempt to eradicate early P. aeruginosa before it became established. I’m sure this would not have been condoned by the regulatory authorities in 2025! However, it should be remembered that at that time, in the early Eighties, desperate measures were needed as CF was usually fatal in childhood or adolescence. Most children suffered from progressive chest infection and most either died during childhood or in their early teens from respiratory failure after years of a miserable chronic illness. There were no adult CF clinics in the early Eighties outside the one at the Royal Brompton in London for the simple reason there were so few adults.

So, at St James’s in Leeds, after discussion with the parents, we started treating a few children with nebulised colomycin 500 mg twice daily after a test dose. The children seemed to tolerate the inhalations. Gradually as more monthly cough/throat swabs became negative, it was quite obvious that the nebulised colomycin was clearing the Ps. aeruginosa from the respiratory tract. At this stage we reported our initial experience in our letter to the Lancet.

The Copenhagen CF centre started first to use inhaled colomycin to reduce relapses in chronic Pseudomonas infection
Jensen T, Pedersen SS, Garne S, Heilmann C, Hoiby N, Koch C. Colistin inhalation therapy in cystic fibrosis patients with chronic aeruginosa lung infection. J Antimicrob Chemother 1987; 19:831-838.Â

The Danish group in Copenhagen, were undoubtedly already the leading CF clinic in the Eighties in Europe (Professor Niels Hoiby and Dr Christian Koch). Apparently following our letter from Leeds to the Lancet reporting its successful use in eradicating early Pseudomonas infection, they started to use nebulised colomycin in the first instance to stabilise the condition of their chronically infected CF patients – suppression rather than eradication.
The Copenhagen CF Centre group trial of colomycin to eradicate early P. aeruginosa
Valerius, NH, Koch, C Hoiby N. Prevention chronic Pseudomonas aeruginosa colonisationin cystic fibrosis by early treatment.~Lane 1991sep 21:338(8769):725-726. https://pubmed.ncbi.nlm.nih.gov/1679870/


Subsequently, after the Jensen trial, apparently as a result of our short letter to the Lancet according to Niels Hoiby, the Copenhagen team performed a controlled trial using nebulised colomycin as early eradication therapy (Valerius et al, 1991). They reported that their results “confirm and extend the preliminary report by Littlewood and colleagues”.
“During the 27 months of the trial,(Valerius et al, 1991) infection with Ps aeruginosa became chronic in significantly fewer treated than untreated subjects (2 [14%] vs 7 [58%]; p <0.05) and there were significantly fewer Ps aeruginosa isolates in routine sputum cultures in the treated group (49/214 [23%] vs 64/158 [41%]; p = 0.0006). Thus, chronic colonisation with Ps aeruginosa can be prevented in cystic fibrosis by early institution of anti-pseudomonas chemotherapy”
The Americans were reluctant to accept the results as historical controls were used which sadly denied US patients this effective treatment for a decade.
As it eventually turned out, both the routine use of nebulised anti-Pseudomonal antibiotics to suppress chronic infection, as recommended by Margaret Hodson in 1981 (also eventually in the USA by Ramsay BW et al, 1999) and the early use of colomycin to eradicate early Pseudomonal infection (Littlewood et al, 1985; Valerius et al, 1991) both proved to be effective, proven and eventually widely used treatments for people with CF on both sides of the Atlantic.
The impressive long term effects of the early treatment on the incidence of chronic P. aeruginosa infection the Leeds CF Centre is described in detail by Tim Lee, now Director of the Leeds Regional Paediatric CF Centre, later in this Paper Trail
Lee TWKeith G Brownlee, Miles Denton, James M Littllewood, Steven Conway. Reduction of chronic Pseudomonas aeruginosa infection at a regional paediatric cystic fibrosis center. Pediatr Pulmonol 2004; 37(12): 104-10. https://pubmed.ncbi.nlm.nih.gov/14730654/
Addendum –
“Pseudomonas aeruginosa eradication: Finally moving the needle?” An editorial by Jonathan Cogen and Margaret Rosenfield of Seattle in the Journal of Cystic Fibrosis 2017; 16:309-310.

“One of the major advances in the treatment of pseudomonas aeruginosa in CF patients over the past 10 to 20 years has been the the widespread adoption of eradication regimens as standard of care for new isolation of P. aeruginosa from respiratory cultures in an effort to delay or prevent chronic P. aeruginosa infection with the associated negative effects on lung health and survival. First pioneered by the Copenhagen CF Clinic in the 1980s, the efficacy of different eradication regimens has been demonstrated in prospective clinic trials, reporting 70-90% eradication rates”.
Thirty five years since the first letter to the Lancet from Leeds reporting early eradication !!!!!!
Conway SP. Miller MG. Ramsden C. Littlewood JM. Intensive treatment of pseudomonas chest infection in cystic fibrosis: a comparison of tobramycin and ticarcillin, and netilmicin and ticarcillin. [ Clinical Trial. Controlled Clinical Trial. Journal Article] Acta Paediatr Scand 1985; 74(1):107-13. https://pubmed.ncbi.nlm.nih.gov/3885674/
Although Steve Conway eventually became an international authority on cystic fibrosis, at this time in 1985 he was my paediatric registrar at St James’s. However, he already had a degree in English and he was and is an excellent and very quick writer! Mike Miller was our first CF Research Fellow.
This is a paper describing the practical details of intensive treatment of Pseudomonas chest infection in CF as developed at our unit; 30 courses of intravenous antibiotics given to 17 children in relapse are described. This was relatively early in the early aggressive intavenous antibiotic treatment of CF. I intended this paper to describe in the detail the various aspects of courses of antibiotic treatment for the paediatricians who intended to treat their patients locally. The aminoglycosides tobramycin or netilmicin were used and the results were comparable. We may have received some funding from one of the firms to do the comparison – I don’t remember. All the authors were involved in the detailed management of the many CF patients and Colin Ramsden, Senior Technician in Microbiology was responsible for the bacteriological investigations; he was a major asset to our unit.
Our comprehensive referral service resulted in our gaining experience in treating some referred children who were in very poor condition. Despite my best intentions not to “take over” such children, approximately 10% of those referred were in such poor condition, as the girl described below, that they were admitted at once to our ward at St James’s for intensive physiotherapy, intravenous antibiotics and nutritional rehabilitation the details of which are described in this present paper. Understandably, some of these families opted to continue their follow-up at St James’s when they observed the often quite dramatic effect of more aggressive antibiotic and nutritional treatment.
The illustrations below are of a referred patient who had been severely under treated. She responded dramatically to three weeks intensive treatment as described in this paper. But the damage was done. Although she continued to respond to similar courses of treatment responses gradually became less impressive and she died aged 16 years.



This was the type of patient referred to as under-treated evident by the dramatic initial response. Unfortunately, although such patients improved quite dramatically initially and for a few years when treated aggressively, they then deteriorated despite treatment (right hand growth chart). In her weight chart the initial improvement is evident with her first course of intravenous anti-pseudomonal antibiotics (lower vertical arrows); subsequent rapid deterioration followed until regular inhaled gentamicin and carbenicillin were introduced. However, although with frequent courses of IV antibiotics and the nebulised antibiotics there was a steady improvement in her weight, eventually her condition gradually deteriorated until she died of respiratory failure aged 16 years.
Two weeks or longer hospital admissions for intravenous antibiotics, intensive physiotherapy and nutritional rehabilitation became regular treatment during the Eighties until our CF Research Fellows and CF Nurse specialist developed home intravenous antibiotic therapy in the late Eighties (described later) after which a proportion were treated at home (Gilbert et al. Arch Dis Child. 1988 May;63(5):512-7. PMID: 3389866 Free PMC article – described later).
Chalmers DM. Brown RC. Miller MG. Clarke PC. Kelleher J. Littlewood JM. Losowsky MS. The influence of long-term cimetidine as an adjuvant to pancreatic enzyme therapy in cystic fibrosis. [ Clinical Trial. Journal Article. Randomized Controlled Trial] Acta Paediatr Scand 1985; 74(1):114-7. https://pubmed.ncbi.nlm.nih.gov/3885675/
Seventeen patients on constant doses of pancreatic enzymes were randomised to receive either cimetidine or placebo for either of two successive six month periods. Nutritional state and maldigestion were assessed at the beginning and end of each period. Reductions in mean values of faecal fat, nitrogen, wet weight, and bile salts of approximately 30% were found on cimetidine therapy. Results showed considerable variation and only the fall in faecal fat (of 33% ) was statistically significant. No benefit was demonstrated for height, weight, skinfold thickness, albumin, vitamin A, bone age or Chrispin-Norman score.
The pancreatic enzymes used at this time (Cotazym 6 to 22 capsules per day) were significantly damaged by the gastric acid reducing their digestive ability. Previous studies were unclear as to whether reducing gastric acid would improve intestinal absorption.
However, this present study was done just as the new acid-resistant microsphere enzymes, Pancrease and Creon, were becoming available. So although we found improved fat absorption in this present study these new acid resistant microsphere preparation soon replaced the older unprotected enzyme powder preparations used in this study (discussed later in Beverley et al, 1987).
Conway SP. Littlewood JM. Admission to hospital with asthma. [ Journal Article] Arch Dis Child 1985; 60(7):636-9. https://pubmed.ncbi.nlm.nih.gov/4026359/
The circumstances surrounding 142 hospital admissions for acute asthma in 110 children during a one year period to our ward at St James’s were examined. Thirty four of 106 (32%) children with previous wheezing had not been diagnosed as asthmatic, nor received effective anti-asthmatic medication. Nineteen of 36 (53%) known, but under-treated, asthmatics were under the care of the hospital paediatricians. Twenty-four of 58 (41%) regular school attenders had missed more than 11 days’ school in the previous year. Good parental understanding of their child’s illness was strongly associated with adequate treatment. Parental understanding was, however, poor in 58 of 137 (42%) admissions. Control of inadequately treated chronic symptoms was obtained by simple and straightforward changes in treatment.
After their admission many of these children were followed up in my outpatient at St James’s where during 1984 we had nearly 342 children attend the asthma clinic at least once. Also I had many asthmatic children referred to the outpatient clinic. The out patient nurses were excellent at doing the childrens’ peak expiratory flow rates, weighing and measuring – their accurate measuring allowed us to observe the slight growth suppressing effect of inhaled corticosteroids (Later reviewed Littlewood, 1988).

I was fortunate to obtain funding for a Specialist Asthma Nurse, Mrs Margaret Graham, who eventually was based in an office in our CF unit. As we had found when we obtained a CF Nurse Specialist, having an experienced nurse involved entirely with one condition allows her/him to accumulate great experience and develop relationships with the children and their families. Margaret remained for many years and we last met in 2019 at Christine Silburn’s (my previous secretary) retirement party in Leeds.